
From Gut Leakiness to Metabolic Resistance
ABSTRACT
The gastrointestinal tract serves as both the body's largest immune organ and a critical barrier between the external environment and systemic circulation. When gut dysbiosis compromises intestinal barrier integrity, bacterial lipopolysaccharide (LPS) translocates from the gut lumen into the bloodstream, triggering a state of chronic metabolic endotoxemia. This LPS-driven inflammation activates Toll-like receptor 4 (TLR4) on immune cells, initiates the NF-κB signaling cascade, and produces a sustained cytokine response (TNF-α, IL-6, IL-1β) that directly causes insulin resistance through IRS-1 serine phosphorylation. This guide presents the complete mechanistic pathway from gut barrier disruption through LPS translocation, immune activation, systemic inflammation, and ultimately metabolic dysfunction. We examine the role of zonulin in tight junction regulation, the TLR4/MyD88/NF-κB signaling axis, tissue-specific metabolic consequences, and evidence-based strategies for restoring gut barrier integrity and breaking the endotoxemia-inflammation-insulin resistance cycle.
TABLE OF CONTENTS
- 1. Introduction: The Gut as a Metabolic Gatekeeper
- 2. Gut Dysbiosis: When the Microbiome Turns Against You
- 3. Intestinal Permeability: The Leaky Gut Mechanism
- 4. LPS Translocation and Metabolic Endotoxemia
- 5. TLR4 Activation and the NF-κB Inflammatory Cascade
- 6. From Gut Leakiness to Systemic Metabolic Disruption
- 7. Tissue-Specific Consequences of LPS-Driven Inflammation
- 8. Clinical Management: Restoring Gut Barrier Integrity
- 9. Clinical Recommendations
- 10. Conclusion
- References
- About the Author
- Intellectual Property and Disclosure
1. Introduction: The Gut as a Metabolic Gatekeeper
The gastrointestinal tract is far more than a digestive organ. It houses approximately 70% of the body's immune cells, contains over 100 trillion microorganisms collectively known as the gut microbiome, and maintains a single-cell-layer barrier that separates the contents of the intestinal lumen from the systemic circulation. When this barrier is intact, it selectively permits absorption of nutrients while preventing the passage of bacterial products, toxins, and undigested food particles into the bloodstream.
When gut barrier integrity is compromised, a condition commonly referred to as "leaky gut" or increased intestinal permeability, the consequences extend far beyond the gastrointestinal tract. Bacterial endotoxins, particularly lipopolysaccharide (LPS) from gram-negative bacteria, gain access to the portal and systemic circulation, triggering a cascade of immune activation and chronic low-grade inflammation that directly drives insulin resistance, metabolic dysfunction, and treatment resistance.
This guide traces the complete mechanistic pathway from gut dysbiosis through barrier disruption, LPS translocation, immune activation, systemic inflammation, and ultimately metabolic resistance. Understanding this pathway reveals why patients with unaddressed gut dysfunction often fail to respond adequately to metabolic interventions, and why gut barrier restoration represents a critical and frequently overlooked component of comprehensive metabolic care.
2. Gut Dysbiosis: When the Microbiome Turns Against You
A healthy gut microbiome exists in a state of dynamic equilibrium, with beneficial bacteria maintaining intestinal barrier integrity, producing short-chain fatty acids (SCFAs), modulating immune responses, and competing with pathogenic organisms. Gut dysbiosis occurs when this equilibrium is disrupted, resulting in a shift toward a pro-inflammatory, barrier-damaging microbial composition.
2.1 Causes of Gut Dysbiosis
- Western diet: High in ultra-processed foods, refined sugars, industrial seed oils, and food additives. Low in fiber, polyphenols, and fermented foods. This dietary pattern starves beneficial bacteria and feeds pathogenic organisms
- Antibiotic use: Broad-spectrum antibiotics indiscriminately eliminate beneficial bacteria, creating ecological niches that pathogenic organisms exploit. Even a single antibiotic course can alter the microbiome for months to years
- Chronic stress: Elevated cortisol alters gut motility, reduces mucosal blood flow, and changes the microbial environment. The gut-brain axis operates bidirectionally, and HPA axis dysregulation directly impacts microbial composition
- Environmental toxins: Pesticides (particularly glyphosate), heavy metals, and endocrine disruptors damage the gut epithelium and alter microbial populations
- Chronic alcohol consumption: Directly damages the intestinal epithelium and promotes the growth of gram-negative bacteria that produce LPS
- Sedentary lifestyle: Physical inactivity is associated with reduced microbial diversity and decreased SCFA production
2.2 Key Microbial Changes in Dysbiosis
Dysbiosis is characterized by several consistent patterns:
- Decreased microbial diversity: Healthy microbiomes are characterized by high species diversity. Dysbiotic states show reduced diversity with dominance by a small number of species
- Loss of Akkermansia muciniphila: This keystone species maintains the mucus layer that protects the intestinal epithelium. Its depletion is consistently associated with increased intestinal permeability and metabolic dysfunction
- Reduced Bifidobacterium and Lactobacillus populations: These genera produce SCFAs, compete with pathogens, and support tight junction integrity
- Increased gram-negative bacteria: Expansion of LPS-producing bacteria (Enterobacteriaceae, Proteobacteria) increases the endotoxin load within the gut lumen
- Decreased butyrate production: Butyrate is the primary energy source for colonocytes and is essential for maintaining tight junction protein expression. Reduced butyrate production directly contributes to barrier breakdown
3. Intestinal Permeability: The Leaky Gut Mechanism
The intestinal barrier consists of a single layer of epithelial cells connected by tight junction protein complexes. These tight junctions are the gatekeepers that control what passes between epithelial cells from the gut lumen into the bloodstream. When tight junction integrity is compromised, the barrier becomes "leaky," allowing paracellular passage of molecules that should be excluded.
3.1 Tight Junction Proteins
The major tight junction protein complexes include:
- Occludin: A transmembrane protein that forms the primary seal between epithelial cells
- Claudins: A family of proteins (claudin-1, -3, -4, -5, -7) that determine the selectivity and permeability characteristics of the tight junction
- Zonula occludens (ZO-1, ZO-2, ZO-3): Scaffolding proteins that anchor transmembrane tight junction proteins to the intracellular actin cytoskeleton
- JAM (junctional adhesion molecules): Support cell-to-cell adhesion and immune cell trafficking
3.2 Zonulin: The Master Regulator of Intestinal Permeability
Zonulin is the only known physiologic regulator of intestinal tight junctions. It is produced by enterocytes in response to specific stimuli and acts by disassembling the tight junction complex, transiently increasing intestinal permeability. Under normal conditions, this is a regulated process. Under pathologic conditions, zonulin overproduction leads to sustained barrier dysfunction.
Triggers of excessive zonulin release include:
- Gliadin (the prolamin component of gluten): Binds to the CXCR3 receptor on enterocytes, triggering MyD88-dependent zonulin release regardless of celiac disease status
- Bacterial overgrowth and dysbiosis: Small intestinal bacterial overgrowth (SIBO) and dysbiotic microbial patterns stimulate zonulin release
- Inflammatory mediators: TNF-α and other cytokines directly upregulate zonulin production, creating a feed-forward loop between inflammation and permeability
3.3 Consequences of Barrier Breakdown
When tight junctions are disassembled by excessive zonulin and inflammatory damage:
- Bacterial LPS gains paracellular access to the lamina propria and portal circulation
- Undigested food antigens cross the barrier, triggering immune responses and food sensitivities
- Bacterial metabolites and toxins enter systemic circulation
- The mucosal immune system is chronically activated, producing sustained inflammatory cytokine output
4. LPS Translocation and Metabolic Endotoxemia
Lipopolysaccharide (LPS) is a structural component of the outer membrane of gram-negative bacteria. It is continuously produced in large quantities within the gut lumen as bacteria proliferate and die. Under normal conditions, the intestinal barrier prevents significant LPS entry into the circulation. When barrier integrity is compromised, LPS translocates into the portal and systemic bloodstream, creating a state of metabolic endotoxemia.
4.1 What Is Metabolic Endotoxemia?
Metabolic endotoxemia, a term coined by Cani et al. (2007), describes a chronic, low-level elevation of circulating LPS that is sufficient to activate innate immune responses and drive metabolic inflammation, but below the threshold that would produce acute sepsis symptoms.
Key characteristics include:
- LPS levels 2 to 3 times higher than healthy baseline (typically measured as endotoxin activity or LPS-binding protein)
- Sustained, chronic elevation rather than acute spikes
- Sufficient to activate TLR4 signaling on immune cells throughout the body
- Associated with insulin resistance, obesity, type 2 diabetes, cardiovascular disease, and NAFLD
4.2 Routes of LPS Entry
- Paracellular translocation: LPS passes between epithelial cells through disrupted tight junctions (the primary route in leaky gut)
- Chylomicron-mediated absorption: LPS can be incorporated into chylomicrons during fat absorption, particularly after high-fat meals. This represents a physiologic route of LPS entry that is amplified by dysbiosis and high-fat diets
- Transcellular transport: Direct passage through damaged or inflamed epithelial cells
Once in the portal circulation, LPS encounters hepatic Kupffer cells (liver-resident macrophages), which serve as the first line of defense. When Kupffer cell clearance capacity is overwhelmed, LPS spills into the systemic circulation, activating immune cells throughout the body.
5. TLR4 Activation and the NF-κB Inflammatory Cascade
Circulating LPS is recognized by the innate immune system through Toll-like receptor 4 (TLR4), a pattern recognition receptor expressed on macrophages, monocytes, dendritic cells, and other immune cells. TLR4 activation by LPS initiates a signaling cascade that culminates in the production of inflammatory cytokines responsible for insulin resistance.
5.1 The TLR4 Signaling Pathway
The LPS-TLR4 signaling cascade proceeds through the following steps:
- LPS binds to LPS-binding protein (LBP) in the circulation, forming an LPS-LBP complex
- The LPS-LBP complex is transferred to CD14, a co-receptor on the surface of macrophages and monocytes
- CD14 presents LPS to the TLR4/MD-2 receptor complex, triggering receptor dimerization and activation
- Activated TLR4 recruits the adaptor protein MyD88 (myeloid differentiation primary response 88)
- MyD88 activates IRAK (interleukin-1 receptor-associated kinase) and TRAF6 (TNF receptor-associated factor 6)
- IRAK/TRAF6 activate the IKK complex (IKKα/IKKβ/NEMO)
- The IKK complex phosphorylates IκB (inhibitor of NF-κB), marking it for proteasomal degradation
- With IκB degraded, NF-κB (p65/p50) translocates to the nucleus
- Nuclear NF-κB activates transcription of pro-inflammatory genes
5.2 NF-κB Target Genes and Cytokine Output
NF-κB activation drives the transcription and release of key inflammatory mediators:
- TNF-α: The primary activator of JNK and IKKβ kinases that cause IRS-1 serine phosphorylation and insulin resistance
- IL-6: Induces hepatic acute-phase response (CRP production) and activates SOCS3, which directly inhibits insulin receptor signaling
- IL-1β: Promotes pancreatic beta-cell dysfunction and amplifies the inflammatory cascade
- IL-18: Contributes to inflammasome activation and sustained inflammatory signaling
- MCP-1 (monocyte chemoattractant protein-1): Recruits additional monocytes/macrophages to sites of inflammation, amplifying the immune response
6. From Gut Leakiness to Systemic Metabolic Disruption
The complete pathway from gut barrier dysfunction to metabolic resistance operates as a sixstep cascade, with each step amplifying the next. Understanding the full pathway reveals the systemic nature of gut-derived metabolic inflammation.
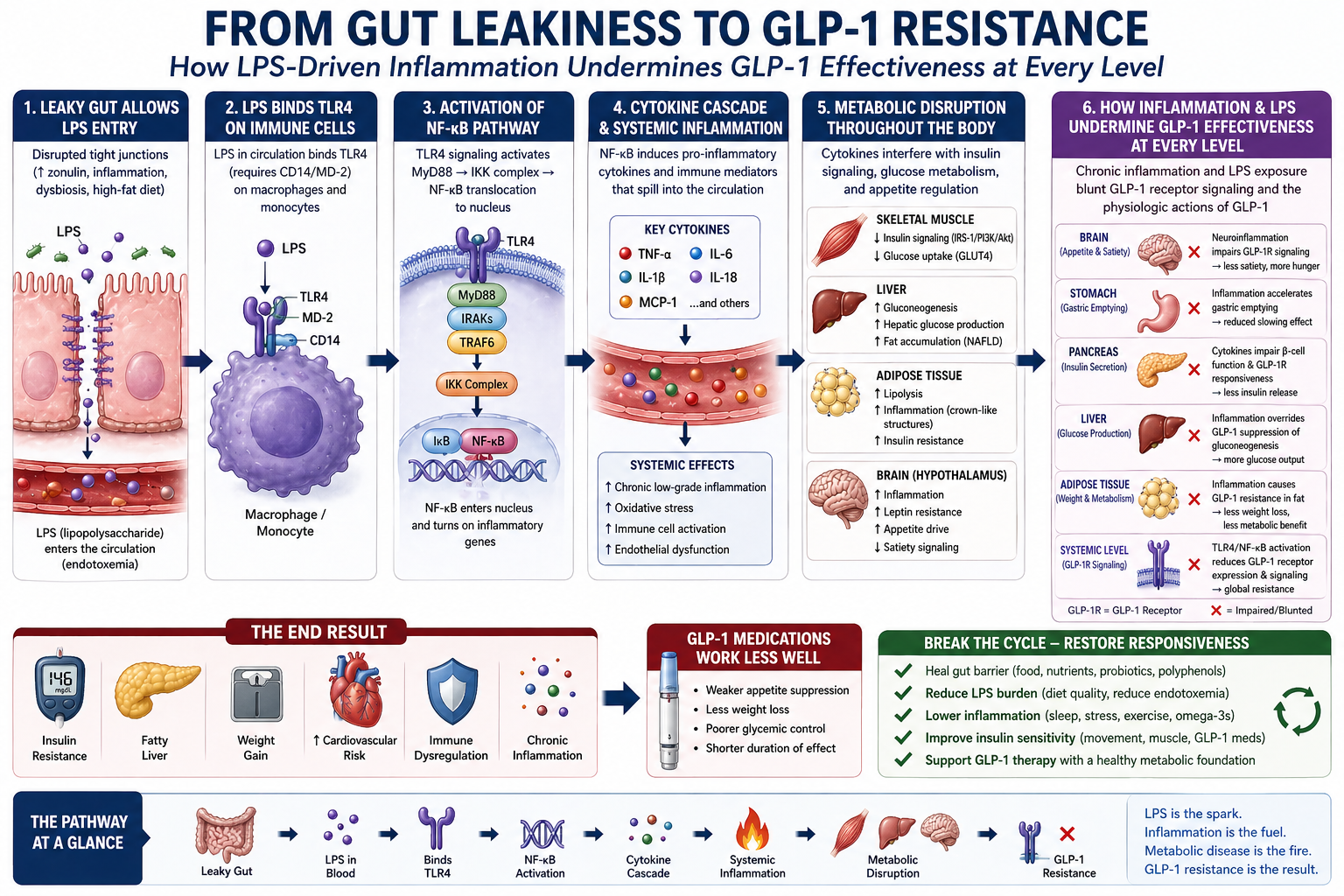
© 2026 Dr. Haad Mahmood, MD. All rights reserved. Original intellectual property. Reproduction prohibited without written permission.
Mahmood H. From Gut Leakiness to GLP-1 Resistance. HaadMD. 2026. https://haadmd.com
6.1 Step 1: Leaky Gut Allows LPS Entry
Disrupted tight junctions, caused by zonulin overproduction, inflammation, dysbiosis, and dietary insults, allow bacterial LPS to cross the intestinal barrier and enter the portal circulation. This is the initiating event in the entire cascade.
6.2 Step 2: LPS Binds TLR4 on Immune Cells
LPS in the circulation binds TLR4 (via the CD14/MD-2 co-receptor complex) on macrophages and monocytes, activating the innate immune response. Hepatic Kupffer cells are the first to encounter portal LPS; when overwhelmed, systemic immune cells are activated throughout the body.
6.3 Step 3: Activation of the NF-κB Pathway
TLR4 signaling activates the MyD88/IRAK/TRAF6/IKK cascade, resulting in NF-κB translocation to the nucleus and activation of pro-inflammatory gene transcription. This converts the immune signal from LPS recognition into sustained cytokine production.
6.4 Step 4: Cytokine Cascade and Systemic Inflammation
NF-κB induces production of TNF-α, IL-6, IL-1β, IL-18, and MCP-1. These cytokines spill into the systemic circulation, creating chronic low-grade inflammation that affects every metabolic tissue in the body. Systemic effects include oxidative stress, immune cell activation, and endothelial dysfunction.
6.5 Step 5: Metabolic Disruption Throughout the Body
The cytokine cascade produces tissue-specific metabolic consequences detailed in Section 7, affecting skeletal muscle, liver, adipose tissue, brain, and pancreas simultaneously.
6.6 Step 6: Treatment Resistance
Chronic inflammation and LPS exposure blunt receptor signaling and impair the physiologic actions of metabolic therapies at every level. Neuroinflammation impairs appetite regulation in the brain. Gastric inflammation accelerates gastric emptying and reduces the slowing effect of satiety therapies. Pancreatic beta-cell inflammation impairs insulin secretion potentiation. Adipose tissue inflammation causes treatment-resistant insulin resistance. The end result is reduced treatment effectiveness across all physiologic targets.
7. Tissue-Specific Consequences of LPS-Driven Inflammation
LPS-driven systemic inflammation produces distinct but interconnected metabolic consequences across the body's major tissues.
7.1 Skeletal Muscle
- Inflammatory cytokines (TNF-α, IL-6) activate JNK and IKKβ kinases, causing IRS-1 serine phosphorylation
- PI3K/Akt pathway inhibition reduces GLUT4 translocation to the cell surface
- Glucose uptake in muscle decreases by 50 to 70% in severely inflamed states
- Impaired glycogen synthesis limits post-exercise glucose storage capacity
7.2 Liver
- Hepatic Kupffer cells are directly activated by portal LPS, producing local TNF-α and IL-6
- Unsuppressed gluconeogenesis increases fasting and postprandial glucose levels
- De novo lipogenesis increases, driving hepatic fat accumulation (NAFLD progressing to NASH)
- Elevated VLDL production increases circulating triglycerides and cardiovascular risk
7.3 Adipose Tissue
- LPS directly activates adipose tissue macrophages, amplifying local inflammation
- Increased lipolysis releases free fatty acids that further impair insulin signaling
- Crown-like structures form around dying adipocytes, creating sustained inflammatory foci
- Insulin resistance in adipose tissue prevents proper lipid storage, driving ectopic fat deposition
7.4 Brain (Hypothalamic Inflammation)
- Systemic cytokines and LPS induce neuroinflammation in the hypothalamus
- Leptin resistance develops, impairing satiety signaling and driving appetite increase
- Dopamine reward circuit dysregulation increases hedonic eating behavior
- Central insulin resistance impairs the brain's regulation of hepatic glucose output
7.5 Pancreas
- IL-1β directly damages pancreatic beta cells, impairing insulin secretion capacity
- Chronic inflammation accelerates beta-cell exhaustion in the setting of sustained insulin resistance
- Impaired incretin potentiation of insulin release reduces the effectiveness of satietybased therapies
8. Clinical Management: Restoring Gut Barrier Integrity
Restoring gut barrier integrity and microbiome balance is essential for reducing systemic endotoxemia and breaking the LPS-inflammation-insulin resistance cycle. Management requires a multi-modal approach targeting the root causes of barrier dysfunction.
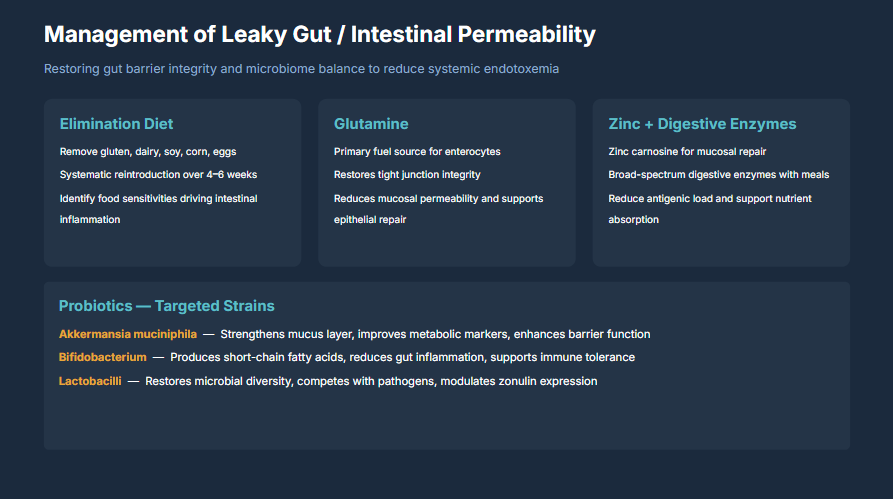
© 2026 Dr. Haad Mahmood, MD. All rights reserved. Original intellectual property. Reproduction prohibited without written permission.
Mahmood H. Management of Leaky Gut / Intestinal Permeability. HaadMD. 2026. https://haadmd.com
8.1 Elimination Diet
The first therapeutic priority is removing dietary triggers that drive intestinal inflammation and permeability:
- Remove gluten, dairy, soy, corn, and eggs for an initial period of 4 to 6 weeks
- Systematic reintroduction of eliminated foods one at a time, monitoring for symptom recurrence, to identify individual food sensitivities
- Identify and eliminate food sensitivities that are driving ongoing intestinal inflammation
- Eliminate ultra-processed foods, refined sugars, and industrial seed oils that feed pathogenic bacteria and damage the epithelium
8.2 Glutamine Supplementation
L-glutamine is the primary fuel source for enterocytes (intestinal epithelial cells) and plays a critical role in barrier restoration:
- Serves as the preferred energy substrate for rapidly dividing enterocytes, supporting epithelial cell turnover and repair
- Directly restores tight junction protein expression, including occludin and ZO-1
- Reduces mucosal permeability and supports epithelial repair in clinical studies
- Typical therapeutic dosing: 5 to 15 grams daily, often divided between meals
8.3 Zinc and Digestive Enzyme Support
- Zinc carnosine: Specifically studied for mucosal repair. Zinc carnosine adheres to the gastric and intestinal mucosa, providing sustained local zinc delivery that supports epithelial healing
- Broad-spectrum digestive enzymes with meals: Improve the completeness of food digestion, reducing the antigenic load presented to the intestinal immune system
- Reduces antigenic load and supports nutrient absorption, ensuring adequate cofactors for barrier protein synthesis
8.4 Targeted Probiotic Strains
Not all probiotics are equivalent. Strain-specific selection based on mechanistic evidence is essential:
- Akkermansia muciniphila: The keystone species for gut barrier integrity. Strengthens the mucus layer, improves metabolic markers (insulin sensitivity, lipid profiles), and enhances barrier function. Available as a pasteurized postbiotic preparation
- Bifidobacterium species (B. longum, B. breve, B. infantis): Produce short-chain fatty acids (particularly acetate and lactate), reduce gut inflammation, and support immune tolerance through regulatory T-cell induction
- Lactobacillus species (L. rhamnosus, L. plantarum, L. reuteri): Restore microbial diversity, compete with pathogenic bacteria for colonization sites, modulate zonulin expression, and support tight junction integrity
8.5 Additional Gut-Supportive Interventions
- Prebiotic fiber: Feeds beneficial bacteria and supports SCFA production. Sources include inulin, fructo-oligosaccharides (FOS), galacto-oligosaccharides (GOS), and resistant starch
- Polyphenols: Compounds from berries, green tea, cocoa, and olive oil support microbial diversity and exert direct anti-inflammatory effects on the gut mucosa
- Bone broth / collagen peptides: Provide glycine, proline, and hydroxyproline that support intestinal mucosal repair
- Butyrate supplementation: Provides the primary fuel for colonocytes when endogenous butyrate production is insufficient due to dysbiosis
9. Clinical Recommendations
- Assess gut barrier function in metabolic patients with treatment resistance. Consider testing for zonulin, LPS-binding protein (LBP), hs-CRP, and calprotectin. Clinical history of food sensitivities, bloating, irregular bowel habits, and antibiotic exposure should raise suspicion for barrier dysfunction.
- Implement an elimination diet as first-line gut intervention. Remove gluten, dairy, soy, corn, and eggs for 4 to 6 weeks with systematic reintroduction. This is both diagnostic and therapeutic.
- Prescribe L-glutamine for barrier restoration. 5 to 15 grams daily to support enterocyte repair and tight junction protein expression.
- Use strain-specific probiotics with mechanistic evidence. Akkermansia muciniphila for barrier integrity and metabolic improvement. Bifidobacterium for SCFA production and immune tolerance. Lactobacillus for microbial diversity and zonulin modulation.
- Supplement with zinc carnosine and digestive enzymes. Zinc carnosine for mucosal repair; broad-spectrum enzymes to reduce antigenic load and support complete digestion.
- Address upstream drivers of dysbiosis. Dietary quality, chronic stress, medication history (particularly antibiotics and PPIs), and environmental toxin exposure must all be evaluated and addressed.
- Monitor inflammatory markers longitudinally. Track hs-CRP, fasting insulin, and HOMAIR at baseline and every 3 to 6 months. Improving inflammation predicts improving metabolic responsiveness.
- Integrate gut barrier restoration into comprehensive metabolic care. Gut health is not a peripheral concern. It is a central determinant of systemic inflammation and treatment responsiveness. Address the gut, and metabolic therapies work more effectively.
10. Conclusion
The pathway from gut leakiness to metabolic resistance is now mechanistically clear. Gut dysbiosis disrupts the intestinal barrier, allowing bacterial LPS to translocate into the systemic circulation. LPS activates TLR4 on immune cells, triggering the MyD88/NF-κB signaling cascade and producing a sustained output of inflammatory cytokines (TNF-α, IL-6, IL-1β). These cytokines cause insulin resistance through IRS-1 serine phosphorylation, disrupt metabolic function across skeletal muscle, liver, adipose tissue, brain, and pancreas, and ultimately impair the effectiveness of metabolic treatments at every physiologic level.
The self-reinforcing nature of this cycle means that without direct intervention at the gut level, metabolic dysfunction will continue to worsen regardless of pharmacologic dose escalation. Inflammation drives insulin resistance, insulin resistance drives further adiposity, adiposity drives further inflammation, and the compromised gut barrier continues to supply the LPS that fuels the entire cycle.
The clinical message is unambiguous: the gut barrier is a metabolic organ. Its integrity determines the inflammatory tone of the entire body, and its dysfunction is a root cause of treatment resistance in metabolic disease. Gut barrier restoration through elimination diet, glutamine supplementation, targeted probiotics, and comprehensive microbiome support is not an adjunctive lifestyle recommendation. It is a foundational therapeutic intervention.
Heal the barrier, silence the inflammation,
and the body remembers how to respond."
~ Dr. Haad Mahmood, MD
References
- Mahmood H. From Gut Leakiness to GLP-1 Resistance [Figure 1]. Correlating GLP-1 Outcomes with Advanced Lab Panels: Cytokines, Metabolic Markers, and Hormones. 2026. Original intellectual property of Dr. Haad Mahmood, MD.
- Mahmood H. Management of Leaky Gut / Intestinal Permeability [Figure 2]. Correlating GLP-1 Outcomes with Advanced Lab Panels. 2026. Original intellectual property of Dr. Haad Mahmood, MD.
- Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761-1772.
- Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470-1481.
- Fasano A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y Acad Sci. 2012;1258(1):25-33.
- Fasano A. All disease begins in the (leaky) gut: role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Res. 2020;9:F1000.
- Depommier C, Everard A, Druart C, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. 2019;25(7):1096-1103.
- Bischoff SC, Barbara G, Buurman W, et al. Intestinal permeability: a new target for disease prevention and therapy. BMC Gastroenterol. 2014;14:189.
- Ghosh SS, Wang J, Yannie PJ, Bhatt DL. Intestinal barrier dysfunction, LPS translocation, and disease development. J Endocr Soc. 2020;4(2):bvz039.
- Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1877-1887.
- Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292(3):E740-E747.
- Derosa G, Maffioli P, Sahebkar A. Improvement of plasma adiponectin, leptin, and C-reactive protein concentrations by orlistat: a systematic review and meta-analysis. Br J Clin Pharmacol. 2016;81(5):828-838.
- Zhou Q, Verne GN. Intestinal hyperpermeability: a gateway to multi-organ failure? J Clin Invest. 2018;128(11):4764-4766.
- Mahmood H. The Physician's Guide to Cortisol Dysfunction in Metabolic Disease. HaadMD Clinical Reference Series. April 2026. Available at: https://haadmd.com
- Mahmood H. How Inflammation Causes Insulin Resistance at the Cellular Level. HaadMD Clinical Reference Series. April 2026. Available at: https://haadmd.com